Our lab, in collaboration with the Professor Robert Vandenberg research group at the University of Sydney (https://www.sydney.edu.au/medicine-health/about/our-people/academic-staff/robert.vandenberg.html), is conducting drug discovery research efforts focused on mitigating the high abuse liability associated with mu-opioid analgesics. Approximately 116 million adults in the US suffer from chronic pain. An estimated 30-40% of these patients experience neuropathic pain. These symptoms include pain induced by non-painful stimuli (allodynia), increased sensitivity to pain (hyperalgesia), and other ongoing pain symptoms. Many neuropathic patients also suffer from comorbidities that reduce quality of life, such as depression, anxiety, or poor sleep quality. Existing pharmacological first-line treatments include gabapentinoids, tricyclic antidepressants, and selective serotonin-norepinephrine reuptake inhibitors, with second- and third-line treatments including opioids. These agents are limited as they provide moderate efficacy, induce dose-limiting side effects, or present a significant risk of tolerance, addiction, abuse, and lethality. Indeed, the dearth of effective therapeutics has led to an overreliance on opioid medications, which has fueled the escalation of opioid use disorder (OUD) incidence, morbidity, and mortality. Furthermore, overuse can also result in opioid-induced hyperalgesia. Thus, the development of new and effective chronic pain therapeutics devoid of the liabilities associated with opioid analgesics remains a highly critical and unmet need. Recent insights into the physiological adaptations underlying chronic pain have encouraged efforts to discover new classes of pain-relieving drugs targeting mechanisms that circumvent mu-opioid receptor engagement.
Among the myriad of cellular and molecular processes identified as contributing to pathological pain, disinhibition of spinal cord nociceptive signaling to higher cortical centers plays a critical role. We, together with others, have demonstrated that impaired glycinergic neurotransmission develops within the dorsal horn of the spinal cord in neuropathic pain models and is an important mechanism underlying mechanical hyperalgesia (amplified pain signaling) and allodynia (a painful response to normally innocuous stimuli). It has been hypothesized that agents capable of augmenting spinal glycinergic neurotransmission may be able to obtund aberrant nociceptor signaling to the brain and serve as a novel class of analgesics. Indeed, drugs that enhance dysfunctional glycinergic transmission in neuropathic pain, and in particular inhibitors of the glycine transporter 2 (GlyT2), are generating widespread interest.
There is compelling evidence that reduced glycinergic signaling in the spinal cord dorsal horn produces mechanical hyperalgesia and allodynia and, importantly, our evidence, and that of others, has established that impaired glycinergic signaling develops in neuropathic states. GlyT2 regulates inhibitory glycinergic neurotransmission via two mechanisms; 1) uptake of synaptic glycine into presynaptic terminals to terminate GlyR signaling, and 2) repackaging glycine into presynaptic vesicles via coordination with the vesicular inhibitory amino acid transporter for subsequent re-release. The sensation of pain can be suppressed by inhibiting GlyT2, which increases glycine concentrations in the synaptic cleft that in turn enhance inhibitory neurotransmission via activation of postsynaptic GlyRs in the spinal cord. Its restricted distribution and high levels of expression in the dorsal horn make GlyT2 an attractive target to augment spinal glycinergic neurotransmission. Changes in spinal glycine levels may affect both tonic neuronal excitability and synaptic glycinergic signaling.
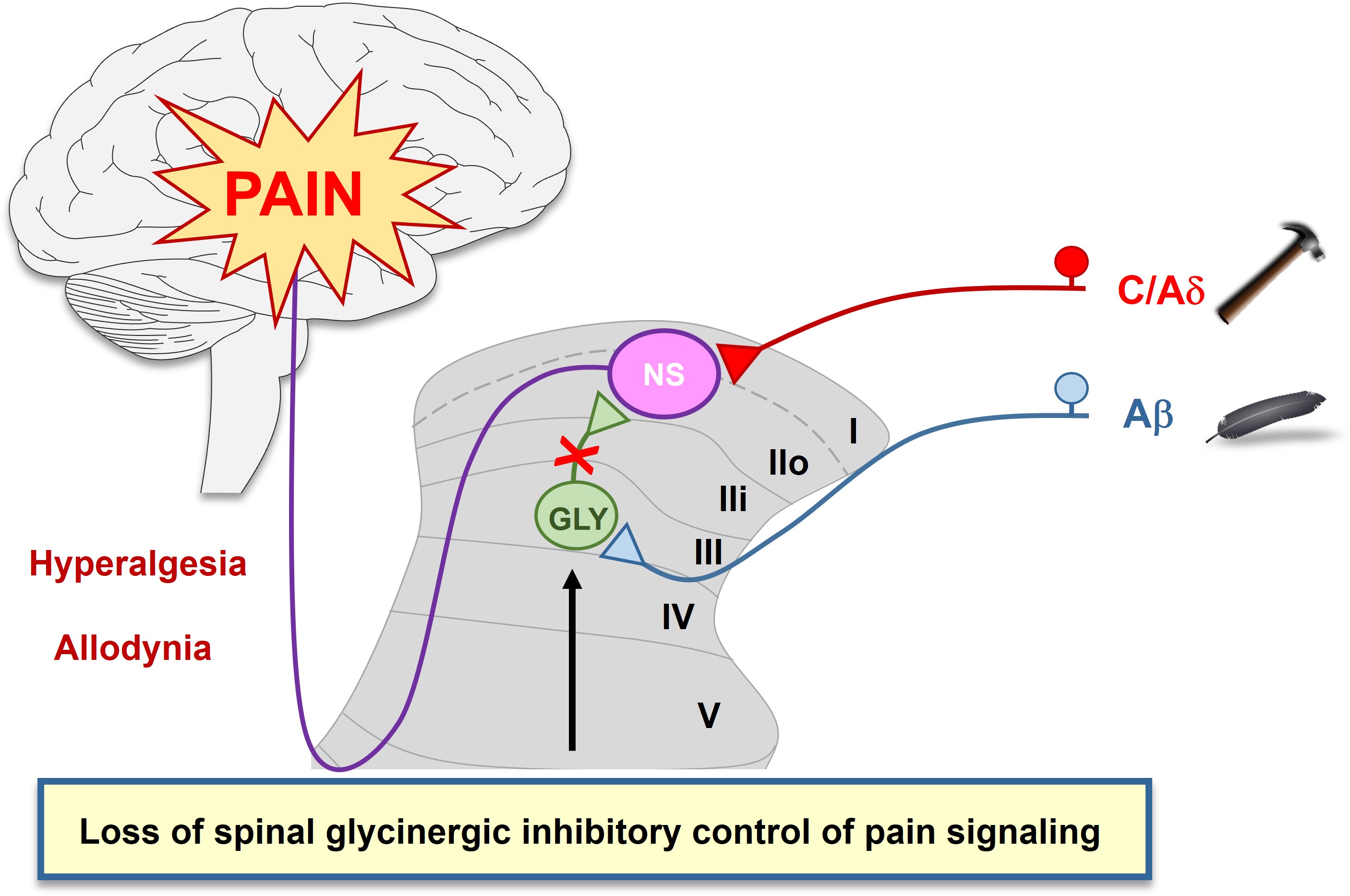
Several GlyT2 inhibitors (see below) have been reported to produce dose-dependent analgesia in models of acute, inflammatory, visceral, cancer, herpetic, and neuropathic pain upon acute and chronic dosing. These findings suggest the development of pain pathologies of varying origin share a reduction of glycinergic inhibitory pain control in the spinal cord. However, a potent, selective, and orally bioavailable GlyT2 inhibitor has yet to reach the clinic.
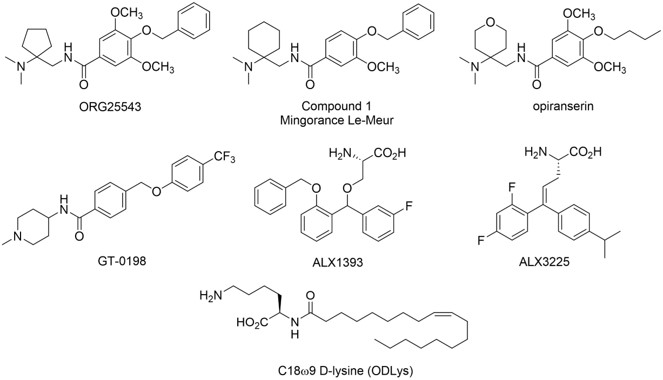
With the assistance of site-directed mutagenesis studies and MD simulations, we are conducting parallel structure-based drug design campaigns to develop non-competitve and competitive inhibitors of GlyT2. Our drug discovery efforts focus on the identification and optimization of novel and orally bioavailable GlyT2 inhibitor preclinical candidates suitable for further development and clinical trials as standalone analgesics or opioid-sparing treatments for neuropathic pain that may help stem OUD.
Funding: NIH 7R01DA048879-04
Cioffi, C. L.; Lotsaris, I.; Cantwell Chater, R. P.; Pati, T. K.; Suleria, K.; Mahesh, G.; Singh, S.; Peiser-Oliver, J.; Mohammadi, S.; Vandenberg, R. J. Current Non-opioid small molecule approaches toward the treatment of neuropathic pain. J. Med. Chem. 2025, 68, 18064−18098.
Cantwell Chater, R. P.; Peiser-Oliver, J.; Pati, T. K.; Quinn, A. S.; Lotsaris, I.; Frangos, Z. J.; Tischer, A. E.; Williams-Noonan, B. J.; O’Mara, M. L.; Michaelides, M.; Mohammadi, S.; Cioffi, C. L.; Vandenberg, R. J.; Shahsavar A. Structural insights into allosteric mechanism of glycine transporter-mediated analgesia. bioRxiv 2025.04.21.649698; doi: https://doi.org/10.1101/2025.04.21.649698.
Gallagher, C. I.; Frangos, Z. J.; Sheipouri, D.; Shimmon, S.; Duman, M-N.; Jayakumar, S.; Cioffi, C. L.; Rawling, T.; Vandenberg, R. J. Development of novel phenylene lipids that are positive allosteric modulators of glycine receptors and inhibitors of glycine transporter 2. ACS Chem. Neurosci. 2023, 14, 2634-2647;PMID: 37466545.
Chater, R. C.; Quinn, A. S.; Wilson, K.; Frangos, Z. J.; Sutton, P.; Jayakumar, S.; Cioffi, C. L.; O'Mara, M. L.; Vandenberg, R. J. The efficacy of the analgesic GlyT2 inhibitor, ORG25543, is determined by two connected allosteric sites. J. Neurochem. 2023. doi: 10.1111/jnc.16028. Epub ahead of print. PMID: 38131125.
Cioffi, C. L. Modulation of glycine-mediated spinal neurotransmission for the treatment of chronic pain. J. Med. Chem. 2018, 61, 2652-2679; PMID: 28876062.
Mostyn, S. N.; Sarker, S.; Muthuraman, P.; Raja, A.; Shimmon, S.; Rawling, T.; Cioffi, C. L.; Vandenberg, R. J. Photoswitchable ORG25543 congener enables optical control of glycine transporter 2. ACS Chem. Neurosci. 2020, 11, 1250-1258; PMID: 32191428.